
一文盘点:单基因糖尿病的研究进展
中国研究型医院学会糖尿病学专业委员会联合医咖会推出:研医论道。针对糖尿病领域的指南、最新文献、会议资讯,学会专家深度解读,全方位剖析领域热点!第19期由天津医科大学总医院的黄雨蒙,舒画和刘铭教授带来『单基因糖尿病研究进展』的精彩综述。
单基因糖尿病是由在胰岛β细胞发育、功能或胰岛素信号通路中起关键作用的单个基因突变导致的一种特殊类型糖尿病,约占所有类型糖尿病的1%-5%[1,2]。按临床症状的发病年龄不同,单基因糖尿病可分为两大类:新生儿糖尿病(NDM)及青少年起病的成人型糖尿病(MODY)。
从广义上讲,线粒体DNA突变导致的线粒体糖尿病,以及由单基因突变引起以糖尿病为临床表现之一的遗传综合征和胰岛素抵抗综合征也可以归属于单基因糖尿病的范畴。
单基因糖尿病临床特征多样,大多数MODY的临床表现与2型糖尿病相似,而由ATP敏感的钾通道(Kcnj11)或胰岛素(Ins)等基因突变导致的NDM的临床表现类似于1型糖尿病。研究显示,有约90%的单基因糖尿病患者都被误诊为1型或2型糖尿病,但是,由于单基因糖尿病的临床预后、转归和治疗方案的选择等,与1型和2型糖尿病有很大差别,目前有近70%患者没有得到正确有效的治疗方案%[3,4]。因此,将单基因糖尿病从1型和2型糖尿病中分离出来,对于提高这些患者的个体化精准治疗至关重要。
随着基因检测技术的提高,未来几年基因筛查成本有望大幅度下降,因此,提高一线临床医生对单基因糖尿病的认识,发现单基因糖尿病特异性临床生物学标志,建立单基因糖尿病临床诊断模型,将是提高单基因糖尿病个体化精准诊治水平的关键。
1. 单基因糖尿病的分型和临床特征
由于突变基因不同,单基因糖尿病临床特征各不相同,目前已发现有至少14种基因的突变可导致MODY,其中,MODY3、MODY2和MODY1最为常见,占所有单基因糖尿病患者的90%以上。
中国研究型医院学会糖尿病学专业委员会联合医咖会推出:研医论道。针对糖尿病领域的指南、最新文献、会议资讯,学会专家深度解读,全方位剖析领域热点!第19期由天津医科大学总医院的黄雨蒙,舒画和刘铭教授带来『单基因糖尿病研究进展』的精彩综述。
单基因糖尿病是由在胰岛β细胞发育、功能或胰岛素信号通路中起关键作用的单个基因突变导致的一种特殊类型糖尿病,约占所有类型糖尿病的1%-5%[1,2]。按临床症状的发病年龄不同,单基因糖尿病可分为两大类:新生儿糖尿病(NDM)及青少年起病的成人型糖尿病(MODY)。
从广义上讲,线粒体DNA突变导致的线粒体糖尿病,以及由单基因突变引起以糖尿病为临床表现之一的遗传综合征和胰岛素抵抗综合征也可以归属于单基因糖尿病的范畴。
单基因糖尿病临床特征多样,大多数MODY的临床表现与2型糖尿病相似,而由ATP敏感的钾通道(Kcnj11)或胰岛素(Ins)等基因突变导致的NDM的临床表现类似于1型糖尿病。研究显示,有约90%的单基因糖尿病患者都被误诊为1型或2型糖尿病,但是,由于单基因糖尿病的临床预后、转归和治疗方案的选择等,与1型和2型糖尿病有很大差别,目前有近70%患者没有得到正确有效的治疗方案%[3,4]。因此,将单基因糖尿病从1型和2型糖尿病中分离出来,对于提高这些患者的个体化精准治疗至关重要。
随着基因检测技术的提高,未来几年基因筛查成本有望大幅度下降,因此,提高一线临床医生对单基因糖尿病的认识,发现单基因糖尿病特异性临床生物学标志,建立单基因糖尿病临床诊断模型,将是提高单基因糖尿病个体化精准诊治水平的关键。
1. 单基因糖尿病的分型和临床特征
由于突变基因不同,单基因糖尿病临床特征各不相同,目前已发现有至少14种基因的突变可导致MODY,其中,MODY3、MODY2和MODY1最为常见,占所有单基因糖尿病患者的90%以上。
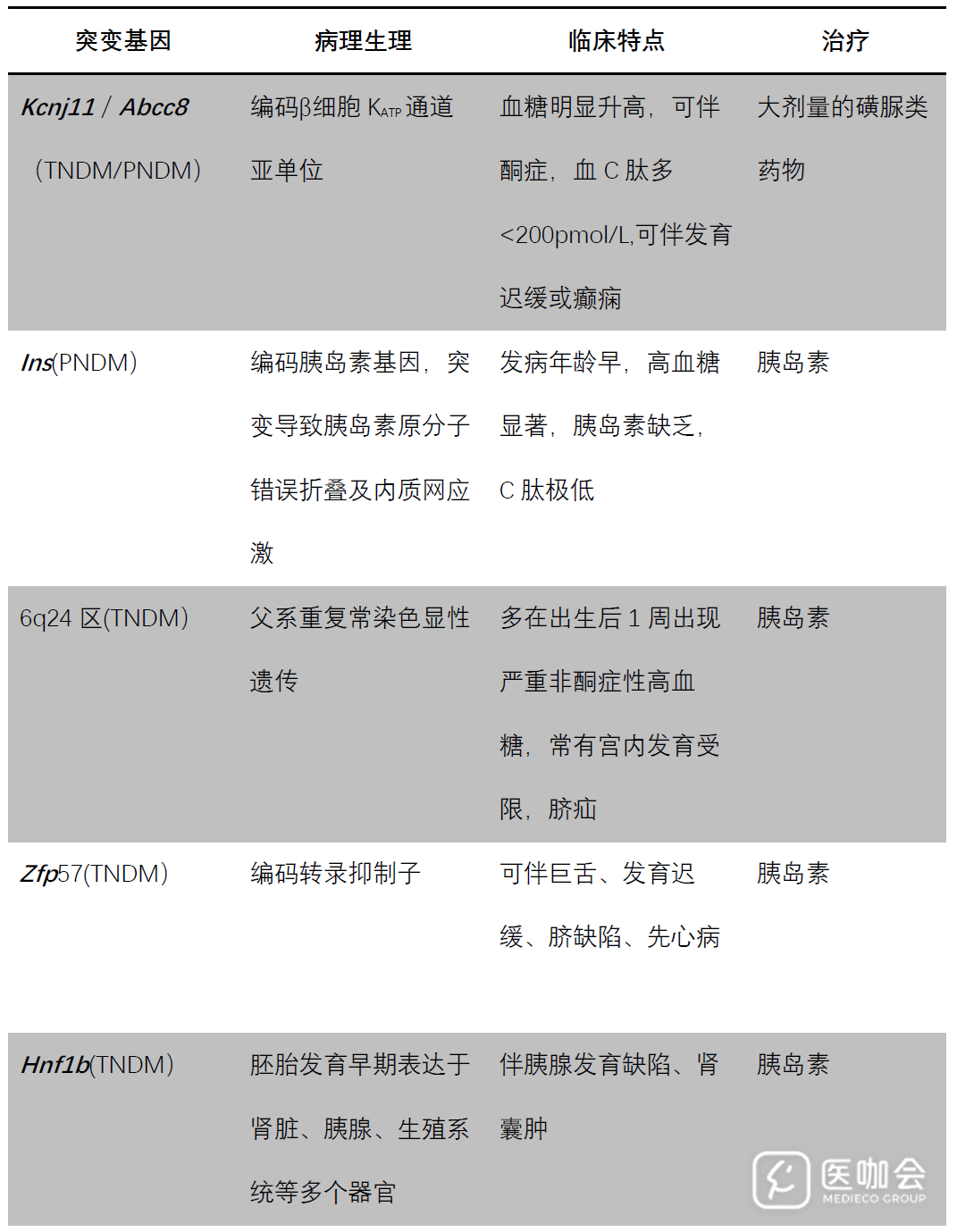
MODY3由肝细胞核因子Hnf1a基因突变引起,是最常见的单基因糖尿病,约占单基因糖尿病总数的50%。Hnf1a广泛表达于胰岛β细胞、肝脏、肠道等器官,是胰岛发育及β细胞分化过程中重要的转录因子,该基因突变导致胰岛功能进行性下降,有较高的外显率,突变基因携带者多在25岁前发病。临床上以餐后血糖明显升高为主,可表现为“三多一少”,但较少发生酮症。
同时,由于Hnf1a是调节肾小管上皮钠葡萄糖共转运体2(SGLT2)表达的重要转录因子,Hnf1a基因突变导致SGLT2表达下降,葡萄糖重吸收减少,肾糖阈降低,故MODY3患者在未发展成糖尿病之前即可出现尿糖阳性。
患病率占第二位的MODY是由葡糖激酶基因(Gck)突变所致的MODY2,葡糖激酶不仅是糖代谢过程中关键的限速酶,在β细胞中它还可以通过感受血糖水平调节胰岛素的分泌,因此被称为“葡萄糖感受器”。MODY2患者多表现为无症状的、非进展性的、轻度空腹血糖升高,糖耐量实验2小时血糖水平仅轻度升高,该类患者并发症风险低,胰岛分泌功能异常不明显[5]。
与MODY3临床特点类似的MODY1型是由Hnf4a杂合突变所致,Hnf4a主要表达于肝脏,但在胰腺和肾脏上也有表达,该基因编码的转录因子通过多种途径影响糖代谢,MODY1患者往往可出现巨大儿和儿童高胰岛素血症,且甘油三脂的水平较低[6]。表1列出常见MODY的突变基因及临床特征。
表1. MODY常见亚型及特点[6,7,8]


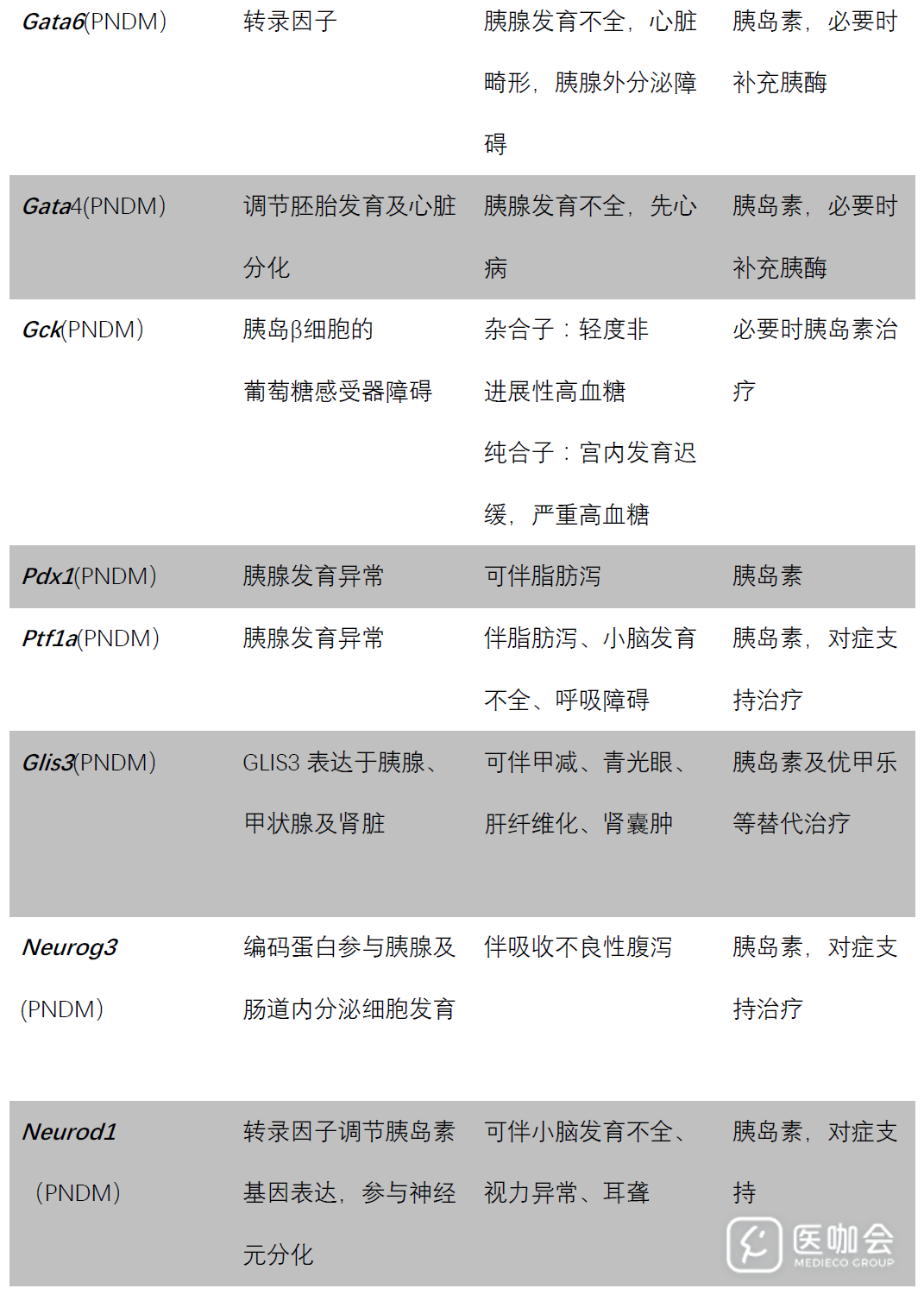
新生儿糖尿病大多发生于出生后6个月以内,也有部分患者推迟至6-12个月发病。其中,约半数以上为永久性 NDM(PNDM),部分NDM可在起病数周至数月后缓解,称为暂时性 NDM(TNDM),约50% TNDM患者在数年至数十年后再次发生糖尿病。
编码在胰岛素分泌中起重要作用的KATP通道Kcnj11/Abcc8基因突变是导致NDM最常见的原因,Kcnj11和Abcc8基因分别编码KATP通道 Kir6.2 亚单位和SUR1调节亚单位,其突变可影响钾离子通道关闭和β细胞膜的去极化、抑制胰岛素分泌,导致常染色体显性遗传性糖尿病。由于Kcnj11也在神经肌肉等组织中表达,约有20%的Kcnj11突变患者可伴发神经系统病变,如发育迟缓、学习障碍、癫痫或肌力下降等。一般认为,Abcc8突变患者发生神经系统病变的概率较低,症状较轻[9]。表2列出常见新生儿糖尿病的突变基因及临床特征。
表2. NDM常见亚型及特点[10,11]

值得注意的是,虽然单基因糖尿病的临床表型主要是由具体突变的基因决定,但是,临床研究发现,同一基因突变或同一位点不同氨基酸的突变所致的糖尿病,在临床特征上有时也可以表现出明显的异质性,如Kcnj11和Ins基因突变既可导致严重的以糖尿病酮症酸中毒为首发症状的新生儿期发病的NDM,也可以导致相对较轻和发病较晚的MODY,这可能与基因本身的功能,不同突变位点对突变蛋白功能的影响,以及突变蛋白对细胞可能造成的“错误蛋白毒性”有关。
除此之外,最近的研究还发现,同一位点相同氨基酸突变也可能表现出不同临床表型,如KCNJ11 C42R突变可以表现为TNDM、儿童期糖尿病甚至在成年后发病[12,13];同样,胰岛素基因突变A24D或C43G发病年龄也可以从新生儿期到10-20岁不等,因此,在2010年,我们在《内分泌内分泌代谢趋势》杂志上,建议将这类糖尿病命名为胰岛素基因突变所致的青少年糖尿病(Mutant Ins-gene induced diabetes of youth, MIDY)[14]。单基因糖尿病临床表型的异质性提示除了单基因突变这个主导的致病因素外,其它的基因和环境因素也可能起一定的作用。
2. 单基因糖尿病相关性遗传综合征
如上所述,部分Kcnj11基因突变的NDM患者合并神经系统病变,其中严重者可合并发育迟缓、早发癫痫、肌无力,被称为DEND(developmental delay,epilepsy,and neonatal diabetes)综合征。这一类特定位点的基因突变不仅影响胰腺内分泌功能,还累及神经和肌肉等多个系统及器官表现出一系列临床综合征,称为单基因糖尿病相关性遗传综合征。
在这类综合征中,目前对编码PERK蛋白的Eif2ak3基因突变所致的Wolcott⁃Rallison综合征研究较为深入。Eif2ak3在人类胰岛和骨组织中高表达,在肾脏、肝脏、胆管等组织中低表达,编码产物为蛋白激酶PERK,其在调节细胞内分泌蛋白的合成和降解、维持内质网蛋白稳态和正常的未折叠蛋白反应中至关重要。
我们和其它研究小组发现Eif2ak3纯合突变或基因敲除小鼠,PERK调节蛋白合成功能出现障碍,导致胰岛素原错误折叠增多并异常堆积于内质网中,诱导持续性内质网应激、细胞凋亡和早发糖尿病[15,16]。
Wolcott⁃Rallison综合征患儿大多在出生后第一个月即出现NDM,伴有特征性的多发性骨骺发育不良及反复发作性的肝肾功能异常。患儿预后不佳,多数在幼年时期死于多器官功能衰竭[17]。
另一种单基因突变所致的遗传综合征——Wolfram综合征(WFS)的发生亦源于调控内质网应激相关蛋白编码基因突变。WFS1蛋白通过影响细胞内钙稳态和未折叠蛋白反应等途径调控内质网应激。Wfs1基因突变导致的Wolfram综合征1型表现为糖尿病、尿崩症、视神经萎缩及耳聋。患儿糖尿病的平均诊断年龄为6岁,随后出现视神经萎缩极易被误诊为糖尿病视网膜病变。
导致Wolfram综合征2型的突变基因Cisd2,编码位于内质网膜调控未折叠蛋白反应、钙离子稳态和自噬的锌指样蛋白,与1型不同,该亚型不出现尿崩症。表3总结了常见的与单基因糖尿病相关的遗传综合征。
表3. 与单基因糖尿病相关的遗传综合征



3. 单基因糖尿病的个体化精准诊治
由于致病基因及发病机制不同,单基因糖尿病的治疗手段需要精准化及个体化,而实现个体化治疗的前提是对单基因糖尿病精确诊断与分型。由于对单基因糖尿病认识不足,并存在与1型或2型糖尿病在临床特征上的相似性,临床上对单基因糖尿病误诊率可达90%,导致患者不能得到最优化的治疗方案,并影响对于疾病进程及预后的判断。
β细胞KATP通道Kir6.2亚单位的编码基因Kcnj11杂合突变是导致PNDM最常见的原因, KATP通道功能异常导致胰岛素分泌减少。由于患儿发病年龄小且易发酮症、C肽水平低,常被误诊为1型糖尿病而接受胰岛素注射治疗。磺脲类药物可与β细胞表面的KATP通道藕联,使此通道关闭,细胞膜去极化,从而释放胰岛素。
发表在新英格兰杂志的研究显示,90%的携带Kcnj11突变患儿可使用磺脲类药物替代胰岛素治疗,且相比胰岛素,高剂量的磺脲类药物可更好控制糖化血红蛋白水平并减小血糖波动,早期确诊可明显提高患者对磺脲类药物的治疗反应(图1)。不仅如此,早期应用磺脲类药物还可以改善Kcnj11突变对神经运动系统的影响,提高患儿的学习能力和神经运动能力[21,22]。
因此,早期正确诊断Kcnj11突变导致的糖尿病,有助于选择最优化的治疗手段,使患者摆脱胰岛素的终身治疗,获得更理想的血糖控制水平,并改善伴发的神经发育异常,极大提高患儿的生活质量和临床预后。
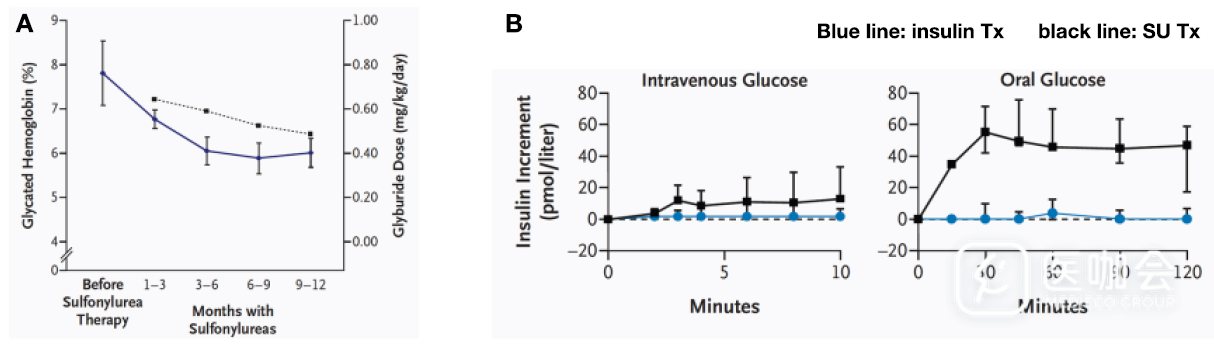
图1. Kcnj11突变所导致糖尿病患者由胰岛素改为磺脲类药物的临床转归。A.磺脲类药物可明显改善患者血糖控制。B.磺脲类药物明显改善患者对葡萄糖刺激的胰岛素分泌反应。(modified from Ewan R.P., etal, NEJM, 2006, 355(5):467-477)
Hnf1a突变导致的MODY3以进行性胰岛素分泌减少为特征,在临床上易被诊断为2型糖尿病而接受二甲双胍降糖治疗。发表于Lancet的一项研究比较了格列齐特和二甲双胍在MODY3和2型糖尿病患者人群中的疗效,结果发现格列齐特对MODY3的降糖效果是二甲双胍的5.2倍,且明显优于格列齐特对2型糖尿病的降糖效果,提示MODY3对磺脲类药物具有高度的敏感性(图2)[23]。目前推荐以低剂量磺脲类药物作为MODY3一线治疗选择。
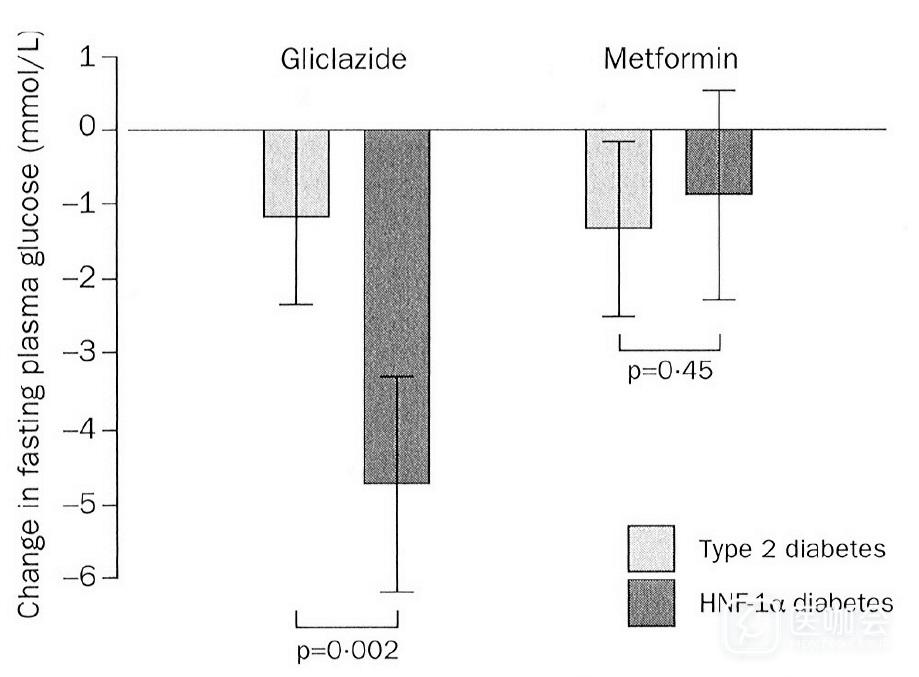
图2. 2型糖尿病和MODY3患者对格列齐特和二甲双胍的治疗反应(摘自Ewan R.P., et al. Lancet. 2003,326 (9392):1275-1281.
Gck基因突变所致的MODY2以轻度、非进展性的空腹血糖升高为特征,在非妊娠期不推荐使用降糖治疗,这是由于多数研究表明不使用降糖治疗并不增加MODY2患者出现糖尿病并发症的风险,因此MODY2患者推荐仅通过单纯饮食、运动控制血糖,并监测糖化血红蛋白,这一发现为MODY2患者摆脱了长期使用降糖药物的心理和经济负担,并极大提高患者的生活质量。
然而,当MODY2患者合并肥胖或胰岛素抵抗,仍有血糖进一步升高并增加发生糖尿病并发症的风险[24]。在妊娠期,部分国家推荐使用胰岛素治疗MODY2以预防胎儿体重过快增长。
综上所述,对单基因糖尿病的诊断和精确分型是实现个体化治疗,给予患者最优化治疗方案的基础,并且对于疾病进程、预后的判断及对家族遗传指导具有重要的意义。
4. 单基因糖尿病与1型和2型糖尿病
1型和2型糖尿病的发生发展是多种基因、环境和生活方式等因素共同作用的结果,同时,1型和2型糖尿病患者个体间也存在的巨大差异,这些都会对致力于研究独立的特定的致病因素对β细胞功能障碍的作用产生影响。
至今为止,全基因组关联分析研究(GWAS)已发现超过150个1型和2型糖尿病的易感基因,在目前已知的可导致单基因糖尿病的30多种基因中,有超过一半的基因多态性与1型或2型糖尿病相关,其中包括Kcnj11/Abcc8、Gck、Hnf1a、Hnf4a, Hnf1b、Pdx1、PPARG等。
因此,尽管从发病机制角度来讲,单基因糖尿病不能完全体现由多种基因和环境因素导致的1型和2型糖尿病的复杂性,但是也正因为其致病原因的单一性,为我们研究独立始动因素对糖尿病发生发展的作用提供了重要的模型。
以胰岛素基因突变为例,至今为止,已有超过50种不同的胰岛素基因杂合突变可以导致人类单基因糖尿病,其中超过70% 的突变都会影响胰岛素前体-胰岛素原在β细胞内质网中的正确折叠过程,导致其错误折叠[25]。
在过去的10年中,我们应用这些突变型的胰岛素原,系统地研究了错误折叠胰岛素原导致胰岛β细胞功能衰竭和糖尿病的分子机制,结果发现突变型胰岛素原不仅自身出现错误折叠,导致在内质网的异常堆积和内质网应激,同时,错误折叠的胰岛素原还可以通过与共表达的野生型胰岛素素原分子间的异常作用,影响野生型胰岛素原的折叠和在细胞内的正常转运和加工,导致有生物活性的成熟胰岛素合成减少和糖尿病的发生[10,26-31]。
更为重要的是,我们应用人和小鼠胰岛的研究发现,在正常β细胞中,有近15-20%新合成的野生型胰岛素原也会出现错误折叠,这些错误折叠的胰岛素原或通过再折叠而达到其正常的空间结构,或通过细胞内的蛋白降解清除系统在细胞内降解,对正常β细胞不会造成影响。
但是,2型糖尿病患者,为应对胰岛素抵抗,β细胞需合成大量的胰岛素原,这就有可能引起错误折叠的胰岛素原增多,当过多的错误折叠的胰岛素原超过β细胞处理能力的阈值时,将引起错误折叠的胰岛素原在细胞内的异常堆积,诱导内质网应激,并加重/导致β细胞功能衰竭[16,25,26,32,33]。
目前有关胰岛素原错误折叠和内质网应激的研究已经成为1型和2型糖尿病研究热点之一,而胰岛素基因突变导致的单基因糖尿病为我们深入理解胰岛素生物合成异常在常见类型糖尿病中的作用及其分子机制提供了重要的“天然模型”。
5. 单基因糖尿病的机遇与挑战
对单基因糖尿病的正确诊断与分型是实现精准个体化治疗的前提条件。分子遗传学检测是诊断单基因糖尿病的金标准,既往通常使用Sanger测序法,该方法准确率高,但成本高、速度慢,逐渐被高通量平行测序的二代测序技术所替代[2]。然而基因检测花费较高,且易遗漏突变基因尚不明确的单基因糖尿病,限制了其在临床上的大规模使用。因此临床上亟需建立简单易操作的单基因糖尿病筛查手段。
美国糖尿病协会2018年发布的糖尿病临床指南特别提出所有6个月以内诊断的糖尿病都应进行新生儿糖尿病的基因检测。另外儿童和成人早发糖尿病,但不具有典型的1型或2型糖尿病特征,并有连续性的早发糖尿病家族史,应做MODY的基因检测[8]。
经典的MODY筛检标准(包括诊断年龄<25岁;糖尿病家族史;自身抗体阴性;非胰岛素依赖等)特异性较高,但敏感性较低,且仅有不到一半的单基因糖尿病患者符合该标准,这意味着使用该筛检标准将漏诊相当一部分MODY患者。
因此,一些学者利用不同的临床特征和实验室参数组合建立筛检模型。近期发表在《Diabetes Care》的一项研究对小于30岁的糖尿病患者,以尿液中C肽/肌酐比值(UCPCR) ≥0.2 nmol/mmol, 且GAD和IA2抗体阴性为为筛检标准,用35种已知的单基因糖尿病亚型对筛检出的可疑患者进行测序,结果发现该模型阳性预测值为20%,即经筛检后1/5的患者为单基因糖尿病,另一方面阴性预测值高达99.9%,提示漏诊率极低[34]。
结合经典的单基因糖尿病筛检模型及近年来对该模型的补充和探索,我们将筛检模型总结如下(图3):

图3. 单基因糖尿病筛检路径模型
a KCNJ11、INS、ABCC8、WSF1等相关基因检测。
b 谷氨酸脱羧酶抗体(GADA)、胰岛细胞抗体(ICA)、胰岛素自身抗体(IAA),酪氨酸磷酸酶抗体(IA2A)、锌转运蛋白-8抗体(ZnT8A) 。
c C肽阳性:C肽>200pmol/L,和/或糖负荷后C肽>200pmol/L,和/或尿C肽/肌酐(UCPRC)>0.2nmol/mol
d MODY2 (GCK),MODY3(HNF1A),MODY1 (HNF4A),MODY5(HNF1B), CEL (MODY 8),PDX1 (MODY4),PAX4 (MODY9),NEUROD1 (MODY6),INS,KCNJ11,ABCC8等相关基因检测。
此外,随机C肽、超敏C反应蛋白、HLA基因型是否可成为单基因糖尿病的生物标记物目前仍处于研究中。Hnf1a调节C反应蛋白的表达,一些研究发现Hnf1a-MODY患者血清中超敏C反应蛋白(hsCRP)的水平显著低于2型糖尿病人群,70-80%的Hnf1a-MODY患者hsCRP蛋白的水平<0.5 mg/l,而仅有20%的2型糖尿病患者达到此标准,这可能与2型糖尿病长期处于慢性低炎症反应的状态有关[35]。
与1型糖尿病迅速衰竭的胰岛功能不同,大多MODY患者在发病数年后往往保留部分胰岛功能,Majidi S.等人以发病年龄小于25岁抗体阴性的糖尿病患者为研究对象,以糖尿病起病6个月以上的随机C肽≥0.15 nmol/L为切点,发现该标准对诊断MODY敏感性可达到83%,阴性预测值为96%,并优于超敏C反应蛋白和高危HLA基因型[36],提示随机C肽可能成为区分MODY与1型糖尿病患者的生物标记物。
对筛检模型的优化和运用特异性的生物学标志物可提高单基因糖尿病的精准诊断,为个体化治疗、疾病进程预后判断和家族遗传指导做好铺垫。然而,由于临床资料相对较少,目前难以找到一种筛检模型或生物学标记物可精确锚定到某种单基因糖尿病的致病基因。提高临床上对单基因糖尿病的认识和完善单基因糖尿病的临床大数据资料,是攻克这一挑战的基础。
参考文献
1. Shepherd, M. et al. Systematic Population Screening, Using Biomarkers and Genetic Testing, Identifies 2.5% of the U.K. Pediatric Diabetes Population With Monogenic Diabetes. Diabetes Care 39, 1879-1888, doi:10.2337/dc16-0645 (2016).
2. Bansal, V. et al. Spectrum of mutations in monogenic diabetes genes identified from high-throughput DNA sequencing of 6888 individuals. BMC medicine 15, 213, doi:10.1186/s12916-017-0977-3 (2017).
3. Yang, Y. & Chan, L. Monogenic Diabetes: What It Teaches Us on the Common Forms of Type 1 and Type 2 Diabetes. Endocrine Reviews 0, er.2015-1116, doi:doi:10.1210/er.2015-1116 (2016).
4. Pihoker, C. et al. Prevalence, Characteristics and Clinical Diagnosis of Maturity Onset Diabetes of the Young Due to Mutations in HNF1A, HNF4A, and Glucokinase: Results From the SEARCH for Diabetes in Youth. The Journal of Clinical Endocrinology & Metabolism 98, 4055-4062, doi:doi:10.1210/jc.2013-1279 (2013).
5. McDonald, T. J. & Ellard, S. Maturity onset diabetes of the young: identification and diagnosis. Annals of clinical biochemistry 50, 403-415, doi:10.1177/0004563213483458 (2013).
6. Anik, A., Catli, G., Abaci, A. & Bober, E. Maturity-onset diabetes of the young (MODY): an update. Journal of pediatric endocrinology & metabolism : JPEM 28, 251-263, doi:10.1515/jpem-2014-0384 (2015).
7. De Franco, E. et al. The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet 386, 957-963, doi:10.1016/S0140-6736(15)60098-8 (2015).
8. American Diabetes, A. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care 41, S13-S27, doi:10.2337/dc18-S002 (2018).
9. Ellard, S. et al. Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects. American journal of human genetics 81, 375-382, doi:10.1086/519174 (2007).
10. Rubio-Cabezas, O. et al. ISPAD Clinical Practice Consensus Guidelines 2014. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatric diabetes 15 Suppl 20, 47-64, doi:10.1111/pedi.12192 (2014).
11. Wen, X. & Yang, Y. Emerging roles of GLIS3 in neonatal diabetes, type 1 and type 2 diabetes. Journal of molecular endocrinology 58, R73-R85, doi:10.1530/JME-16-0232 (2017).
12. Yorifuji, T. et al. The C42R mutation in the Kir6.2 (KCNJ11) gene as a cause of transient neonatal diabetes, childhood diabetes, or later-onset, apparently type 2 diabetes mellitus. J Clin Endocrinol Metab 90, 3174-3178, doi:10.1210/jc.2005-0096 (2005).
13. Liu, L. et al. Mutations in KCNJ11 are associated with the development of autosomal dominant, early-onset type 2 diabetes. Diabetologia 56, 2609-2618, doi:10.1007/s00125-013-3031-9 (2013).
14. Liu, M. et al. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends in Endocrinology & Metabolism 21, 652-659, doi:10.1016/j.tem.2010.07.001 (2010).
15. Gupta, S., McGrath, B. & Cavener, D. R. PERK (EIF2AK3) Regulates Proinsulin Trafficking and Quality Control in the Secretory Pathway. Diabetes 59, 1937-1947, doi:10.2337/db09-1064 (2010).
16. Liu, M., Li, Y., Cavener, D. & Arvan, P. Proinsulin disulfide maturation and misfolding in the endoplasmic reticulum. J Biol Chem 280, 13209-13212 (2005).
17. Deeb, A., Al-Zidgali, F. & Ofoegbu, B. N. Multicystic dysplastic kidney: a new association of Wolcott-Rallison syndrome. Endocrinology, diabetes & metabolism case reports 2017, doi:10.1530/EDM-17-0090 (2017).
18. Habeb, A. M. et al. Pharmacogenomics in diabetes: outcomes of thiamine therapy in TRMA syndrome. Diabetologia, doi:10.1007/s00125-018-4554-x (2018).
19. Senniappan, S. et al. Pigmentary hypertrichosis and non-autoimmune insulin-dependent diabetes mellitus (PHID) syndrome is associated with severe chronic inflammation and cardiomyopathy, and represents a new monogenic autoinflammatory syndrome. Journal of pediatric endocrinology & metabolism : JPEM 26, 877-882, doi:10.1515/jpem-2013-0062 (2013).
20. Marshall, J. D. et al. Alstrom Syndrome: Mutation Spectrum of ALMS1. Human mutation 36, 660-668, doi:10.1002/humu.22796 (2015).
21. Pearson, E. R. et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. The New England journal of medicine 355, 467-477, doi:10.1056/NEJMoa061759 (2006).
22. Beltrand, J. et al. Sulfonylurea Therapy Benefits Neurological and Psychomotor Functions in Patients With Neonatal Diabetes Owing to Potassium Channel Mutations. Diabetes Care 38, 2033-2041, doi:10.2337/dc15-0837 (2015).
23. Pearson, E. R. et al. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 362, 1275-1281, doi:10.1016/S0140-6736(03)14571-0 (2003).
24. Owen, K. R. Treating young adults with type 2 diabetes or monogenic diabetes. Best practice & research. Clinical endocrinology & metabolism 30, 455-467, doi:10.1016/j.beem.2016.05.002 (2016).
25. Liu, M. et al. INS-gene mutations: From genetics and beta cell biology to clinical disease. Mol Aspects Med 42, 3-18, doi:10.1016/j.mam.2014.12.001 (2015).
26. Guo, H. et al. Inefficient translocation of preproinsulin contributes to pancreatic beta cell failure and late-onset diabetes. J Biol Chem 289, 16290-16302, doi:10.1074/jbc.M114.562355 (2014).
27. Wright, J. et al. Dominant protein interactions that influence the pathogenesis of conformational diseases. The Journal of Clinical Investigation 123, 3124-3134, doi:10.1172/jci67260 (2013).
28. Liu, M. et al. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes 61, 828-837, doi:10.2337/db11-0878 (2012).
29. Liu, M. et al. Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport. PLoS One 5, e13333, doi:10.1371/journal.pone.0013333 [doi] (2010).
30. Hodish, I. et al. Misfolded Proinsulin Affects Bystander Proinsulin in Neonatal Diabetes. Journal of Biological Chemistry 285, 685-694, doi:Doi 10.1074/Jbc.M109.038042 (2010).
31. Liu, M., Hodish, I., Rhodes, C. J. & Arvan, P. Proinsulin maturation, misfolding, and proteotoxicity. Proceedings of the National Academy of Sciences 104, 15841-15846, doi:10.1073/pnas.0702697104 (2007).
32. Liu, M., Wright, J., Guo, H., Xiong, Y. & Arvan, P. in Vitamins & Hormones Vol. Volume 95 (ed Litwack Gerald) 35-62 (Academic Press, 2014).
33. Scheuner, D. et al. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med 11, 757-764, doi:10.1038/nm1259 (2005).
34. Shields, B. M. et al. Population-Based Assessment of a Biomarker-Based Screening Pathway to Aid Diagnosis of Monogenic Diabetes in Young-Onset Patients. Diabetes Care 40, 1017-1025, doi:10.2337/dc17-0224 (2017).
35. Owen, K. R. Monogenic diabetes: old and new approaches to diagnosis. Clinical medicine 13, 278-281, doi:10.7861/clinmedicine.13-3-278 (2013).
36. Majidi, S. et al. Can Biomarkers Help Target Maturity-Onset Diabetes of the Young Genetic Testing in Antibody-Negative Diabetes? Diabetes technology & therapeutics 20, 106-112, doi:10.1089/dia.2017.0317 (2018).



